
Amyloïdose neurologie
Erfelijke ATTR amyloïdose
Erfelijke ATTR amyloïdose in het kort
hATTR of familiale transthyretine amyloïdose is het gevolg van een (autosomaal dominant) erfelijke puntmutatie van het gen van het precursor eiwit transthyretine (TTR) dat wordt geproduceerd in de lever. Door deze puntmutatie is het eiwit TTR minder stabiel en kan het opstapelen in verschillende plekken in het lichaam. Met behulp van DNA-onderzoek kan de ziekte aan het licht worden gebracht. Eerstegraads familieleden van iemand met erfelijke ATTR amyloïdose hebben 50 procent kans dat zij de mutatie van het gen ook hebben. De meest voorkomende vorm is de V50M-mutatie (voorheen beschreven als V30M), waarbij valine vervangen wordt door methionine op positie 30/50 van het transthyretine eiwit.1
Epidemiologie
hATTR amyloïdose is uiterst zeldzaam. De geschatte incidentie in Nederland is 3 per jaar (100 patiënten in 33 jaar, van 1985 tot 2018), de geschatte prevalentie 3 per miljoen (nu circa 50 onder controle bij 17 miljoen mensen). De ziekte treft evenveel mannen als vrouwen.2
In bepaalde landen komt amyloïdose met polyneuropathie (hATTR-PN) vaak voor zoals Portugal, Zweden, Cyprus, Mallorca en Japan. Men schat in Noord-Portugal bijvoorbeeld dat zelfs 1 op 538 mensen de ziekte hebben.Amyloïdose met cardiomyopathie (hATTR-CM) komt frequenter voor bij de Afro-Amerikaanse bevolking.1, 3
hATTR is een onomkeerbare, progressieve autosomaal dominante aandoening. Dat maakt de aandoening vrij regionaal gebonden. Echter dwingt de hedendaagse migratietendens tot oplettendheid van elke arts en is een grondige familiale anamnese aan te raden bij het stellen van de diagnose. 4, 5
Symptomen en klachten
Er zijn twee belangrijke fenotypes te onderscheiden volgens de symptomen die zich manifesteren. Welke symptomen zich eerst manifesteren hangt erg af van welke mutatie:
- Men spreekt van hATTR met cardiomyopathie (hATTR-CM) als er cardiale symptomen zijn.
- Men spreekt van hATTR met polyneuropathy (hATTR-PN) als er sensomotorische disfuncties zijn.
Ook een gemengd fenotype is mogelijk.1
De belangrijkste symptomen en klachten kunnen betrekking hebben op:
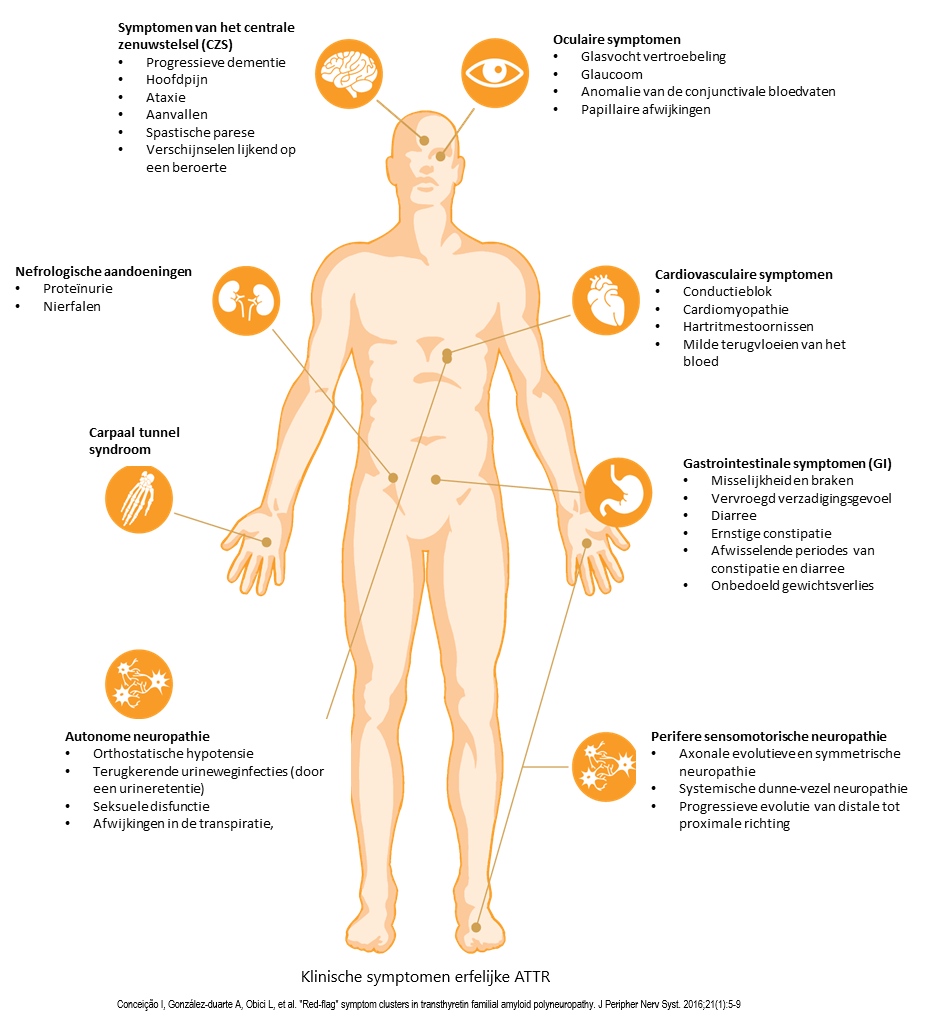
Autonome symptomen ontwikkelen zich gewoonlijk in de vroege stadia van de ziekte, vóór het begin van een motorische beperking of een algemene verslechtering van de kwaliteit van leven van de patiënt.5 Deze symptomen kunnen variëren afhankelijk van het type mutatie. De vroege autonome stoornissen zijn vooral refractaire constipatie, alternerende constipatie en diarree of enkel diarree, verlies van eetlust en gewichtsverlies, misselijkheid en erectiestoornissen. In een later stadium ontwikkelt de patiënt ook andere autonome stoornissen zoals urineproblemen (moeilijk urineren, urineretentie of incontinentie) en orthostatische hypotensie.1
Ziekteverloop
De mediane overleving bedraagt ongeveer 10 jaar.6
Gezien het onomkeerbare progressieve karakter van de aandoening is een vroegtijdige diagnose uiterst belangrijk in de hoop de ziekte enigszins te stabiliseren. Maar de brede waaier aan symptomen zijn doorgaans verantwoordelijk voor een soms jarenlange doorverwijzing met foutieve diagnoses en behandelingen als gevolg. Dit kan belangrijke gevolgen hebben omdat de mediane leeftijd van de patiënt op het moment van de diagnose 63 jaar is en zijn levensverwachting 2 tot 15 jaar is vanaf het begin van de symptomen.4, 5
Diagnose
Het is van belang om de ziekte zo snel mogelijk op te sporen om daarmee de ziekte in een zo vroeg mogelijke fase te stabiliseren en verdere progressie te vertragen. Hiermee is de verwachting dat niet alleen de overleving verbetert, maar ook de ziektelast zo laag mogelijk gehouden wordt. hATTR is een autosomaal dominant erfelijke ziekte waarbij ook familieleden geholpen kunnen worden met een diagnose.2
Verschillende specialismen kunnen een rol spelen bij het identificeren van deze patiënten. De neuroloog en pijnspecialisten kunnen deze vervullen bij patiënten met tekenen van een beginnende polyneuropathie of autonome neuropathie die niet direct kan worden verklaard. Hierbij moet de drempel laag liggen om (naast vrije lichte ketens-, NT-proBNP- en urineanalyse) via de genetica DNA-diagnostiek te overwegen. De cardioloog kan dit doen bij patiënten met tekenen van onverklaard hartfalen bij een verdikt myocard. Hierbij moet de drempel laag liggen om (naast vrije lichte ketens- en urineanalyse) via de genetica DNA-diagnostiek te overwegen, naast het verrichten van een bisfosfonaat-botscan om verhoogde tracer-opname in het hart op te sporen. De internist of maag-darmarts kan dit doen bij patiënten met diarree en gewichtsverlies dat in eerste instantie niet kan worden verklaard. Vaak vindt een scopie gelijk plaats met het verrichten van een biopsie. Belangrijk dat bij de aanvraag ook naar de aanwezigheid van amyloïd wordt gevraagd, zodat het biopt ook wordt gekleurd met Congorood. De oogarts wordt bezocht bij visusproblemen door glasvochttroebelingen. Hierbij moet de drempel laag liggen om via de genetica DNA-diagnostiek te overwegen.2
Hebt u een patiënt met bepaalde neurologische en/of cardiale klachten, die mogelijk ook gepaard gaan met autonome of andere symptomen, hou dan zeker hATTR in uw achterhoofd en refereer uw patiënt tijdig naar één van de erkende referentiecentra.
Onderstaande figuur kan helpen om de meest voorkomende symptomen te identificeren:

Diagnostisch algoritme Cardiologische symptomen bij ATTR amyloïdose
Diagnostisch algoritme Neurologische symptomen bij ATTR amyloïdose
Erfelijkheidsonderzoek
Om vast te stellen of sprake is van een erfelijke vorm van ATTR amyloïdose is DNA-onderzoek geïndiceerd. Eerstegraads familieleden van iemand met erfelijke ATTR amyloïdose hebben 50 procent kans dat zij de mutatie in het gen ook hebben.1
Wanneer genetisch onderzoek een genmutatie heeft aangetoond, is doorverwijzen naar een klinisch geneticus aanbevolen voor counseling en advies bij eventueel familieonderzoek. Door de behandelaars in het expertisecentrum wordt nadrukkelijk geadviseerd om familieleden die boven de 18 zijn te verwijzen naar klinische genetica in hun eigen buurt voor genetisch onderzoek. De aanwezigheid van een mutatie kan daar worden vastgesteld en, indien aanwezig, ook ethische zaken besproken zoals preconceptieadvies en het overwegen van in-vitrofertilisatie met pre-implantatie diagnostiek en embryoselectie.2
Bekijk hier de Richtlijn informeren van familieleden bij erfelijke aandoeningen.
Naast de opsporing van asymptomatische dragers van een mutatie is tijdige detectie van mensen met deze ziekte noodzakelijk. Van belang is dat de familie in zo breed mogelijke zin wordt geïnformeerd, zodat (achter-)neven, (achter-)nichten en familieleden die nog verder weg zijn, op de hoogte zijn dat de erfelijke ziekte in hun familie voorkomt. Helaas is dit een grijs gebied waar privacy en bevoegdheden van genetica en familie dwars door elkaar heen lopen. Wellicht kan hier een rol voor de patiënten, vertegenwoordigd via de SAN (Stichting Amyloïdose Nederland), weggelegd zijn.2
Bekijk de onderstaande talkshow ATTR amyloïdose & Erfelijkheid voor meer informatie, of beluister de podcast over het belang van genetisch testen.
Ziektemechanisme7,8
- TTR wordt hoofdzakelijk in de lever aangemaakt en circuleert gewoonlijk als een tetrameer van vier gevouwen eiwitsubeenheden (monomeren).
- Mutaties in het TTR-gen zijn aangetoond als oorzaak voor ATTR-polyneuropathie.
- Er zijn meer dan 100 verschillende mutaties van het TTR-gen gerapporteerd. De meest voorkomende mutatie is V50M (voorheen ook genoemd V30M).
- Mutaties veranderen de volgorde van aminozuren en produceren zo monomeren die de vrije tetrameer destabiliseren.
- Destabilisatie resulteert in dissociatie van het tetrameer in gevouwen monomeren, die vervolgens verkeerd vouwen en aggregeren wat uiteindelijk cumuleert in het ontstaan van amyloïdfibrillen.
- Amyloïdfibrillen slaan neer in perifere en autonome zenuwen en in andere organen, zoals het hart, ogen, maagdarmkanaal en nieren.
Behandeling
Naast de behandeling van de aangedane organen en ziekteverschijnselen verschillend per patiënt, kan fysiotherapie, diëtiek, psychologie of maatschappelijk werk een onderdeel uitmaken van deze behandeling. Multidisciplinaire aanpak is essentieel.
Afhankelijk van type en het stadium van de ziekte is ook een directe behandeling van erfelijke amyloïdose met medicatie mogelijk. Zoals de behandeling waarbij het transthyretine-eiwit met medicijnen wordt gestabiliseerd. Daardoor wordt minder snel amyloïd gevormd. De behandeling van erfelijke ATTR kan ook worden gedaan met behulp van het zogeheten gene-silencing (gen-deactivatie).9

Ga voor meer informatie over de behandeling van amyloïdose naar de website van het expertisecentrum Amyloïdose van het UMCG: www.amyloid.nl, of neem contact op met de expertisecentra Amyloïdose van het UMCG en het UMC Utrecht.


Bronnen
1. Adams D. et al. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol 2016, 29 (suppl 1):S14 - S26.
2. Richtlijn diagnostiek en behandeling van erfelijke ATTR amyloïdose GRaCE. https://www.amyloid.nl/wp-content/uploads/2019/12/Richtlijn-ATTRm-amyloidose-versie-3.pdf
3. Maurer M. et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016;68(2):161-172.
4. Swiecicki P. et al. Hereditary ATTR amyloidosis: a single-institution experience with 266 patients. Amyloid. 2015;22(2):123-131.
5. Gonzalez-Duarte A. Autonomic involvement in hereditary transthyretin amyloidosis (hATTR amyloidosis). Clin Auton Res. 2019;29(2):245-251.
6. Stichting Amyloïdose Nederland (SAN), de Vereniging Samenwerkende Ouder- en Patiëntenorganisaties (VSOP) en het Nederlands Huisartsen Genootschap (NHG). Informatie voor de huisarts over amyloïdose. Soest 2016.
7. Benson M. et al. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve. 2007;36:411-423.
8. Hou X. et al. Transthyretin and familial amyloidotic polyneuropathy: recent progress in understanding the molecular mechanism of neurodegeneration. FEBS J. 2007;274:1637-1650
9. Nativi-Nicolau J. et al. Amyloidosis cardiomyopathy: update in the diagnosis and treatment of the most common types. Curr Opin Cardiol. 2018 Sep;33(5):571-579
Ook interessant
-
‘In veertig jaar tijd was ik niet één dag ziek…’
‘‘Ik hoop dat cardiologen en andere medisch specialisten mijn verhaal lezen en daardoor snel aan de bel trekken bij patiënten zoals ik.’’ Wim de Ket (74) weet sinds een jaar dat hij de zeldzame aandoening transthyretine gerelateerde cardiale amyloïdose heeft. Hij prijst zichzelf gelukkig dat de diagnose binnen enkele maanden nadat hij zich met klachten bij de huisarts meldde al werd gesteld. ‘‘Ik spreek genoeg mensen die jaren moesten wachten op een diagnose. Dat is funest bij deze ziekte.’’

