Amyloïdose neurologie
AL amyloïdose
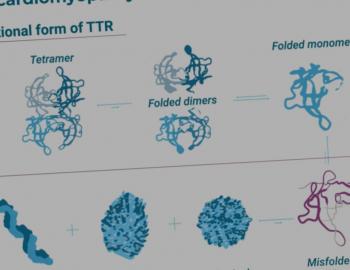
Systemische amyloid light chain (AL) amyloïdose ontstaat in het beenmerg. Een groep plasmacellen – dit zijn de zogenaamde voorlopercellen van kankerplasmacellen – klontert daar samen tot een zogenaamde ‘kloon’. De plasmacellen maken losse lichte ketens van eiwitten aan. Deze eiwitten zien er anders uit dan normaal en zijn verkeerd gevouwen. Daardoor blijven ze als het ware in organen en weefsels van het lichaam aan elkaar vastplakken en stapelen zich op nadat ze daar via het bloed zijn gekomen. AL amyloïdose is verwant aan andere plasmacelziektes zoals het Multipel Myeloom (ziekte van Kahler) en MGUS ('monoklonale gammopathie van onbekende betekenis').1
Epidemiologie
In Nederland heeft AL amyloïdose een incidentie van ongeveer 10 patiënten per miljoen inwoners per jaar. De gemiddelde leeftijd waarop AL amyloïdose zich manifesteert ligt tussen de 55 en 60 jaar. Het lijkt erop dat mannen iets vaker de ziekte krijgen dan vrouwen.2
Symptomen en klachten
De symptomen en klachten van AL zijn zeer uiteenlopend waardoor het lastig is om de juiste diagnose te stellen. Bij dit type amyloïdose kan vrijwel in ieder orgaan en weefsel amyloïd gestapeld worden. De symptomen en klachten kunnen zijn cardiomyopathie, vergrote organen, proteïnurie en nierfunctiestoornissen, ernstige diarree en neuropathie. Onderstaand figuur geeft de klinische symptomen van AL amyloïdose weer:
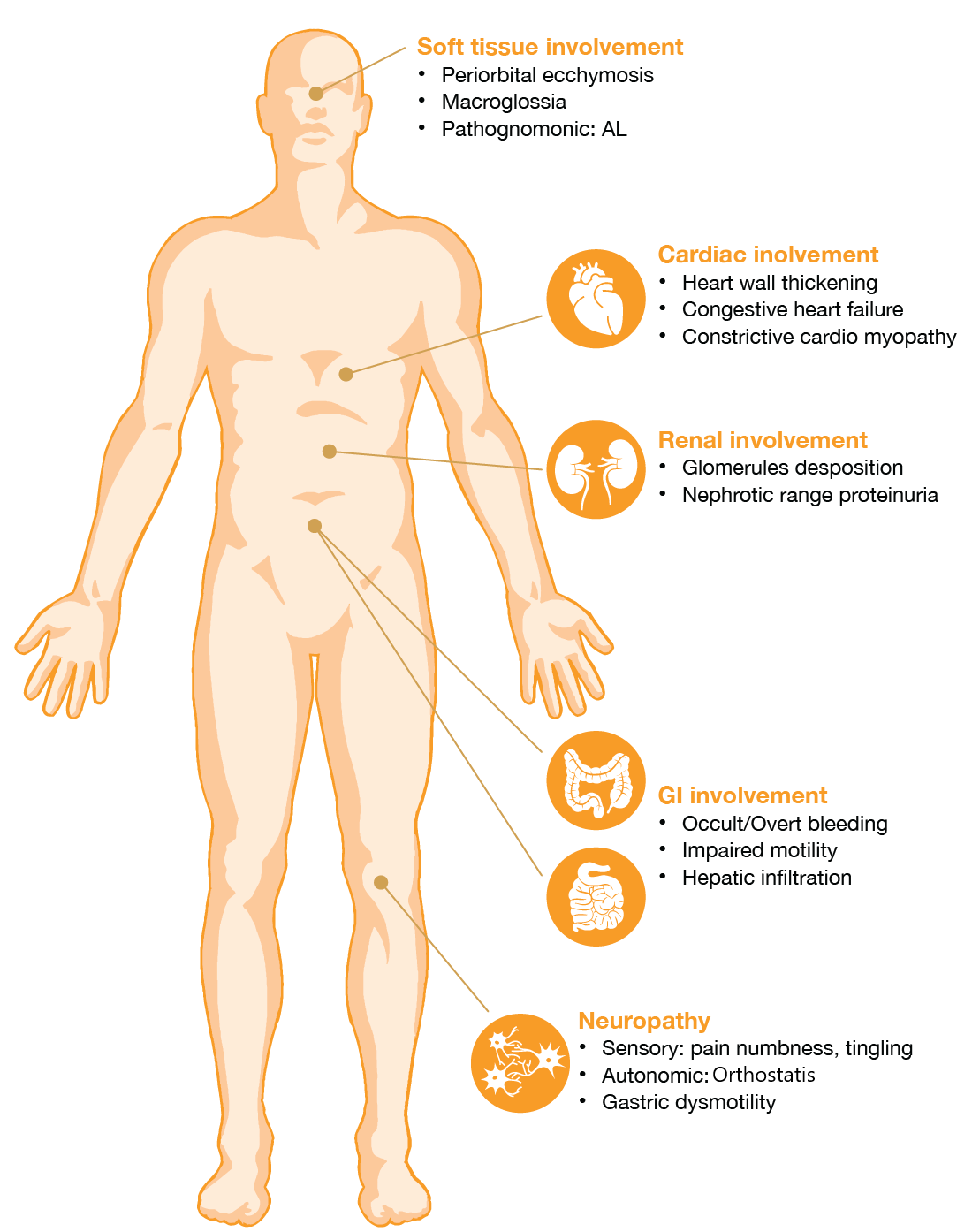
Klinische kenmerken die specifiek zijn voor AL amyloïdose zijn een combinatie van macroglossie en periorbitale purpura. Deze doen zich echter alleen maar voor bij minder dan een derde van alle gevallen. Betrokkenheid van zachte weefsels, buiten het carpaletunnelsyndroom, is nagenoeg uniek voor AL amyloïdose. Macroglossie, musculaire pseudohypertrofie en vergroting van de speekselklieren komen vaak voor.3
Ongeveer een vijfde van alle patiënten met systemische AL amyloïdose hebben perifere neuropathie bij presentatie. Geïsoleerde neuropathie bij afwezigheid van andere orgaanbetrokkenheid komt niet vaak voor bij AL amyloïdose.3
Cardiale betrokkenheid speelt ook een grote rol bij AL amyloïdose. Zo wordt stapeling van amyloïd in het hart bij ca. 50% van de patiënten gezien.4
Ziekteverloop
Wanneer AL amyloïdose patiënten niet worden behandeld, bedraagt de gemiddelde overleving 12-18 maanden. De mate van betrokkenheid van het hart is de belangrijkste prognostische parameter. Is er eveneens sprake van ernstig hartfalen, dan bedraagt de overleving slechts 3-4 maanden. Het probleem bij deze specifieke patiëntenpopulatie is, dat zij een zware behandeling meestal niet aankunnen omdat hun lichamelijke conditie (zeer) slecht is. Daarnaast duurt het enige tijd voor het effect van de behandeling optreedt en die tijd hebben ze soms gewoonweg niet. Behandelde patiënten bij wie de therapie aanslaat, hebben een verbeterde overleving die (mediaan) kan oplopen tot ongeveer 5 jaar.5
Diagnose
Snelle diagnostiek en behandeling van AL amyloïdose is essentieel voor het behoud van orgaanfuncties en het is van cruciaal belang reeds in een vroeg stadium een hematoloog, cardioloog en, indien nodig, een neuroloog en nefroloog te betrekken.
Bij een verdenking op AL amyloïdose moet altijd eerst met een labonderzoek worden getest op de aanwezigheid van monoklonaal eiwit om de diagnose AL-amyloïdose te bevestigen en/of te verwerpen.6 Hiernaast kunnen uitslagen als serum albumine sediment, kreatinine klaring en proteinurie belangrijke aanwijzingen geven. De diagnose kan worden gesteld door het afnemen van een biopt; als daarin amyloïd wordt gezien, wordt de diagnose daarmee doorgaans bevestigd. Belangrijk dat bij de aanvraag naar de aanwezigheid van amyloïd wordt gevraagd, zodat het biopt ook wordt gekleurd met Congorood.5
Voor meer informatie over de diagnose, sleutelindicatoren en een diagnostisch algoritme kijk op:
Behandeling
De behandeling van AL amyloïdose is gericht op het behandelen van de achterliggende gestoorde gezondheid van de plasmacel, waardoor de abnormaal verhoogde spiegel van de lichte keten eiwitten daalt. De plasmacellen worden vernietigd door middel van chemotherapie, indien mogelijk gevolgd door een transplantatie van eigen stamcellen. De medicijnen die worden gebruikt voor de behandeling van AL amyloïdose zijn gelijk aan die van de behandeling bij het Multipel Myeloom maar worden gebruikt in andere doseringen en schema’s. Geadviseerd wordt om de behandeling over te laten aan een expertisecentrum.7,8
Ga voor meer informatie over de behandeling van AL amyloïdose naar de website van het expertisecentrum van het UMC Utrecht of het expertisecentrum Amyloïdose van het UMCG.

Bronnen
1. https://www.amyloidose.nl/wat-amyloidose/al-amyloidose-systemisch
2. https://iknl.nl/nieuws/2019/al-amyloidose-als-ziektebeeld-in-de-nederlandse-ka
3. Wechalekar A. et al. Systemic amyloidosis. Lancet. 2016 Jun;25;387(10038):2641-54.
4. Merlini G. CyBorD: stellar response rates in AL amyloidosis. Blood. 2012;119:4343–45
5. Stichting Amyloïdose Nederland (SAN), de Vereniging Samenwerkende Ouder- en Patiëntenorganisaties (VSOP) en het Nederlands Huisartsen Genootschap (NHG). Informatie voor de huisarts over amyloïdose. Soest 2016 https://vsop.nl/media/magazine/huisartsenbrochure-amyloidose/files/assets/basic-html/page-1.html#
6. Oerlemans, M, et al. Cardiac amyloidosis: the need for early diagnosis. Neth Heart J. 2019;27, 525–536
7. Zicht op Zeldzaam. Kwaliteitsstandaard AL-Amyloïdose https://zichtopzeldzaam.nl/documenten/amyloidose-kwaliteitsstandaard/
8. https://www.amyloid.nl/behandeling/#behandelingsmogelijkheden-pro
Ook interessant
-
‘Ik was vooral opgelucht toen ik de diagnose kreeg’
Het was Peter zelf die ontdekte dat hij AL amyloïdose heeft. Hij moest aandringen in het ziekenhuis voordat zijn vermoeden werd onderzocht én bevestigd. ‘‘Ik was vooral opgelucht toen ik de diagnose kreeg. Eindelijk wist ik wat ik had. En kon ik beginnen met een behandeling, voordat het te laat was.’’ De zestiger vertelt over de periode voor zijn diagnose, zijn behandeltraject en hoe het nu met hem gaat.
-
Neurologische uiting van ATTR amyloïdose
Gezondheidswinst door een snelle diagnose
Differentiaaldiagnose | Klinische presentatie | Diagnose | Behandeling
In het begin sluimeren de symptomen. Pas na gemiddeld 4 jaar kondigt de neurologische vorm van ATTR amyloïdose (ATTR-PN) zich aan: de patiënten krijgen symptomen van polyneuropathie. Vanaf dat moment is de levensverwachting, zonder behandeling, gemiddeld 6 tot 12 jaar.1